
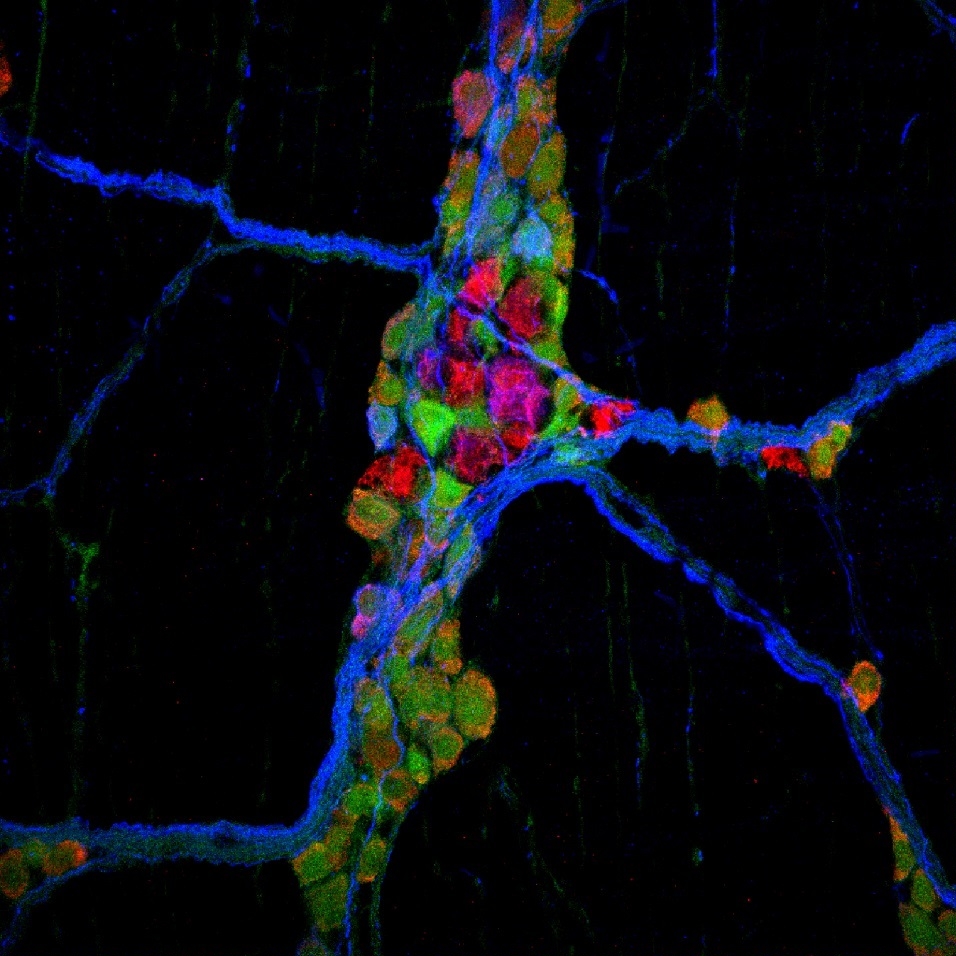
Hirschsprung disease (HSCR) is a congenital disorder caused by genetic mutations that prevent normal innervation of the rectum and distal colon. In severe cases, longer segments of colon and even portions of the small intestine are also affected. This defective innervation causes the bowel’s muscle to contract tightly, thus causing obstruction and preventing normal passage of stool. Infants affected by this disorder can also develop severe inflammation of the colon and small bowel known as enterocolitis. HSCR is treated by surgical resection of the aganglionic bowel. Our lab uses animal models and tissues obtained from HSCR patients to understand the pathophysiology of this disease. We have shown that the absence of enteric neurons and glial cells in HSCR does not only cause abnormal gut motility but also leads to abnormalities in the intestinal epithelium, smooth muscle, and microbiota. Although HSCR most visibly impacts the distal large bowel, we have uncovered evidence that the entire gastrointestinal tract functions abnormally even after corrective surgery. We are studying RNA expression and epigenetics in animal models to understand why these grossly normal portions of the intestine function improperly. We are also applying our models and clinical samples to understand why HSCR patients develop severe inflammation. Insights from this work will help us develop novel, less invasive therapies to treat infants with this devastating disease.